
A 67-year-old man with head and neck cancer who received radiation treatment two weeks earlier was admitted to a palliative care center for treatment of severe pain caused by a massive fungal infection in and around his throat. At the time of his admission, his pain had been treated for one week (with partial success) with a 50 μg/hr fentanyl patch. Shortly after admission, he was given itraconazole, an antifungal drug, to treat the infection. The next day signs of opioid toxicity abruptly appeared. The hospital staff replaced the fentanyl with morphine, the opioid toxicity abated, and the patient was symptom-free two days later. He was later released with a half-dose (25 μg/hr) fentanyl patch, which satisfactorily relieved his pain without causing opioid overdose-like symptoms.
Source: JPSM VOLUME 24, ISSUE 3, P284-286, SEPTEMBER 01, 2002
https://doi.org/10.1016/S0885-3924(02)00477-3
What happened?
It's well known that blood levels of some drugs can be affected by the presence of a second drug (also by foods and beverages) resulting in a phenomenon called drug-drug interactions. The name is somewhat misleading because the drugs do not actually interact directly with each other. Instead, this effect is caused by drug #2 inhibiting (1) the liver enzymes responsible for the metabolism of drug #1, resulting in higher (2) blood levels and a longer duration of action. There are numerous databases of known drug-drug interactions and it is important that doctors and pharmacists provide this information to patients to avoid what happened in the case study above.
Some questions to ponder:
- Was it a coincidence that the patient developed opioid toxicity following the initiation of antifungal therapy?
- Why did this toxicity cease when fentanyl was replaced by morphine?
- Why was the lower dose patch suddenly the "correct" dose when a higher dose had been used earlier?
Hint: Some opioids are "boosted" by the presence of other drugs, while others are not. Believe it or not, this all makes sense, so hang in there.
Metabolism: Two types for the price of one
The liver is the primary "processing center" in our bodies; most of the enzymes responsible for drug and chemical metabolism are found there. But there are two distinct types of metabolism that are as different as night and day in terms of which molecules they process and the way they do it.
Phase I Metabolism – CYPs modify or degrade drugs
The liver is very "clever." It can tell which chemicals/drugs need to be broken down and eliminated and the best way to do so, using one or both metabolic pathways. The first step is called Phase I metabolism – a "heavy-duty" process where large and lipophilic (water-insoluble) molecules are modified by being degraded into smaller fragments, oxidized (typically by replacing a hydrogen atom with a hydroxyl (OH) group), or both. Here is an example of two Phase I oxidation reactions of fentanyl, both of which are promoted by a family of enzymes called cytochrome P-450 or CYPs (2) (Figure 1).
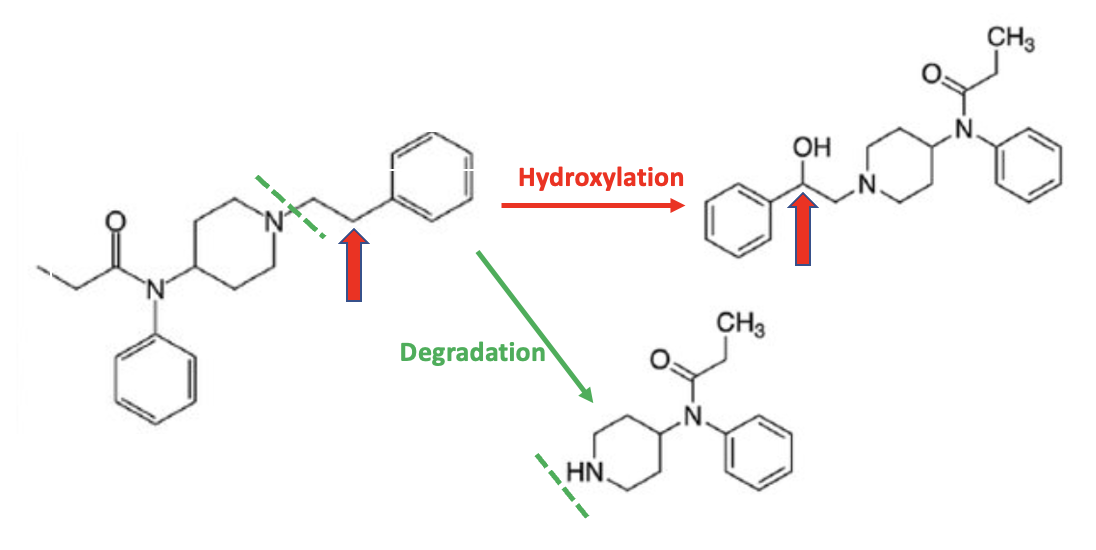
Figure 1. Two examples of Phase I metabolism of fentanyl are given in Figure 1. The top reaction is an example of a hydroxylation reaction, where the carbon atom indicated by the red arrow loses a hydrogen atom and gains a hydroxyl group (red arrow). The bottom reaction is a degradation; part of the molecule is removed, as indicated by the green hatched line. Both reactions are catalyzed by CYP450 enzymes in the liver.
Phase II Metabolism - UGTs promote drug elimination in the urine
As discussed in the previous paragraph, the liver selects drugs that need to be degraded or oxidized prior to elimination (Phase I metabolism). But it also recognizes that different types of drugs are already set up to be excreted, albeit with a minor modification. These drugs still need a "nudge" to make them more water-soluble; this is the purpose of Phase II metabolism. A water-solubilizing group, commonly glucuronic acid, is attached to a hydroxyl (OH) group) to form what is called a "glucuronide conjugate." Once the conjugate is formed the molecule becomes highly water soluble and makes its way to the urine via the kidneys. Phase II metabolism is carried out by a different family of liver enzymes called UDP-glucuronosyltransferases, mercifully abbreviated as UGTs.
It should now be clear (more or less) why a primary function of Phase I CYPs is to add a hydroxyl group to the drug or chemical. The hydroxyl group acts as a "handle" that the UGTs use to attach glucuronic acid. This is how the two enzymes work together by sequentially adding a hydroxyl group (Phase I) and then making the metabolite water soluble – a two-step process sequentially carried out by two very different liver enzymes. Below is an example of the Phase II reaction of morphine (Figure 2). Note that morphine already contains two hydroxyl groups. As a result, the molecule is neither hydroxylated nor broken down. Instead, morphine itself (as its glucuronide conjugate) is excreted otherwise unchanged.
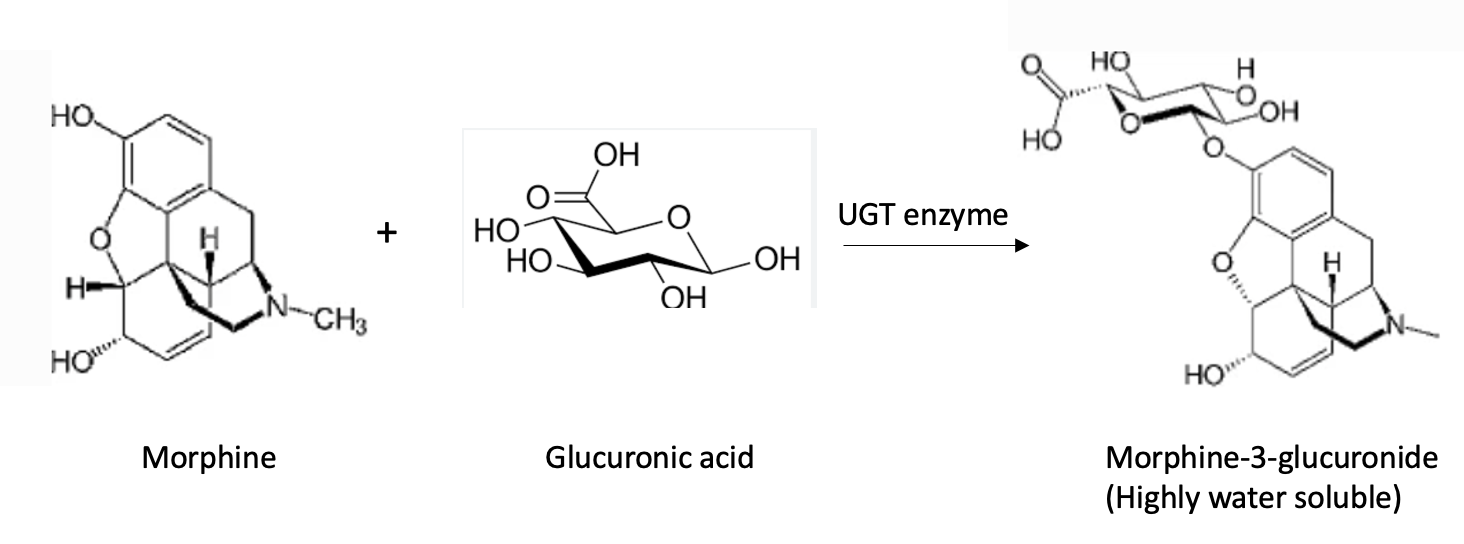
Figure 2. The reaction of morphine with glucuronic acid (which the body makes from glucose) is catalyzed by a UGT enzyme. The resulting product, morphine-3-glucuronide, is extremely water-soluble (polar, hydrophilic) and is excreted in the urine. Note that this reaction is the simple addition of a solubilizing group to one of the hydroxyl groups on morphine, not an oxidation or degradation reaction; those steps, if necessary, are first performed by Phase I enzymes.
How does this explain the differences between fentanyl and morphine in the case report?
There is a marked difference in how the liver treats fentanyl and morphine. Fentanyl is broken down by CYP3A4 (do NOT ask about the name), one of the two most important members of the CYP family. Guess what drug is a powerful inhibitor of CYP3A4? The antifungal drug itraconazole. It inhibits ("ties up") CYP3A4 so effectively that it is less available to normally metabolize fentanyl. Now, let's answer the questions I posed above.
1. Was it a coincidence that opioid toxicity developed following the initiation of itraconazole therapy?
Absolutely not. Symptoms of fentanyl overdose only began once itraconazole therapy was initiated. And we now know why – the fentanyl metabolism was partially blocked because the itraconazole inhibited the enzyme that would have normally metabolized it. The result was abnormally high levels of fentanyl.
2. Why did this toxicity cease when morphine replaced fentanyl?
Morphine is not metabolized by CYP3A4; its metabolism was not affected by the itraconazole (See Figure 3, below). This is why opioid toxicity ceased when fentanyl was replaced by morphine. In other words, CYP3A4 is not responsible for the metabolism of morphine, so its inhibition by itraconazole won't affect the levels of morphine.
3. Why was the 25 μg/hr patch all of a sudden the "correct" dose when 50 μg/hr had been used earlier?
To prepare the patient for discharge his team switched him from IV morphine back to the fentanyl patch but also cut his fentanyl dose by half to account for the drug-drug interaction caused by Itraconazole, which was still being administered (the minimum time to treat oral fungal infections is at least three weeks). When the patient was discharged (and still taking Itraconazole) his pain relief, which was barely adequate at 50 μg/hr, was satisfactory at the half-dose and without any observed opioid toxicity. Part of this, of course, was that the fungal infection, which was very painful, was resolved.
It would be difficult to come up with a better example of drug-drug interaction than what you've just read.
What about other opioids?
You can now (maybe) answer this for yourself by looking at the chemical structures of some commonly-used opioids.
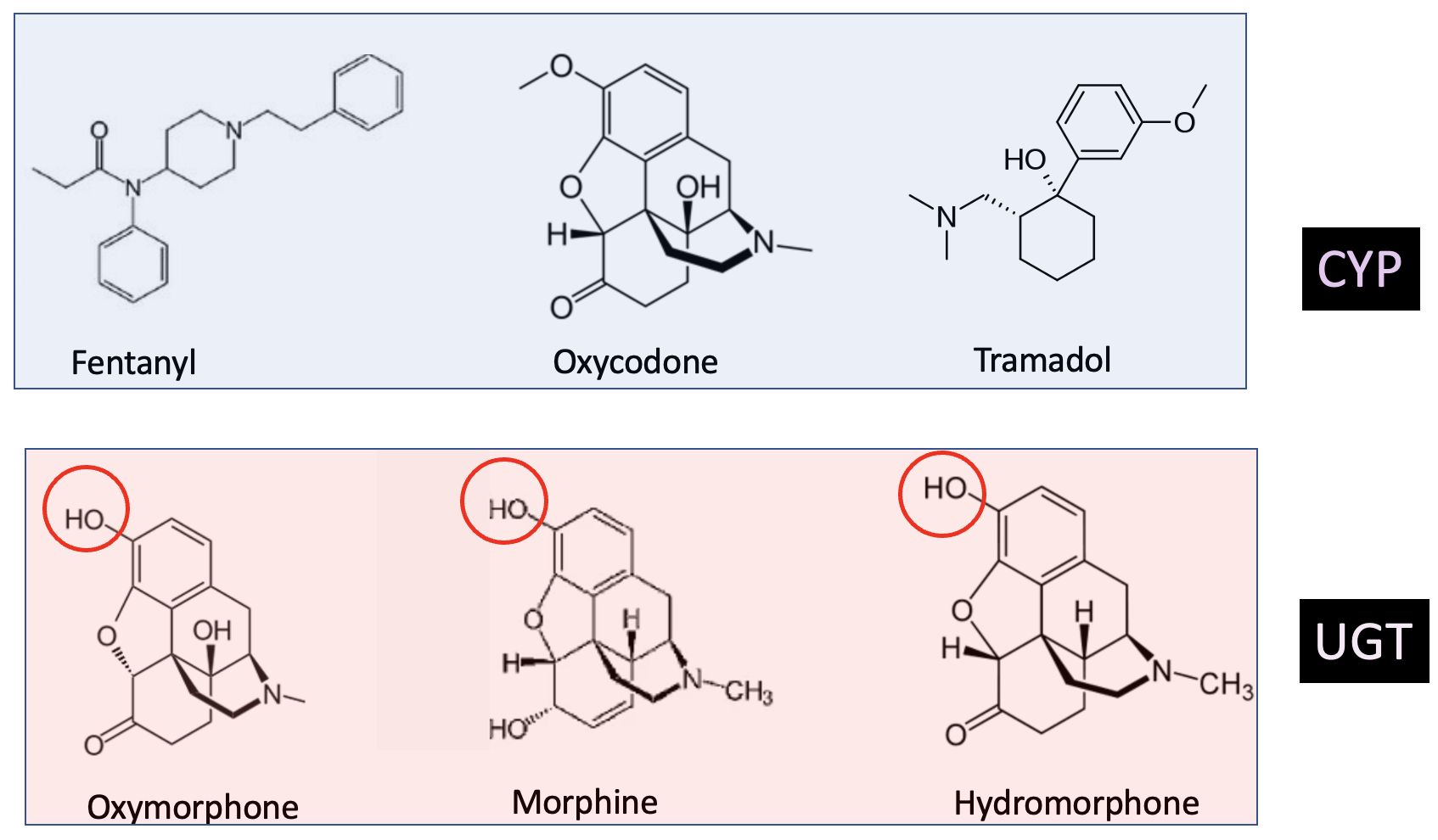
Figure 3. The three drugs in the light blue box are all metabolized by CYP3A4, which means that a person taking fentanyl, oxycodone, or tramadol would have to be aware of the potential for elevated opioid blood levels (this should also be stated on the pill bottle). The three drugs in the pink box are metabolized by UGT enzymes due to the presence of the hydroxyl groups (red circles) and are not affected by drugs like Itraconazole. It's rather astounding that two drugs as similar as structurally similar as oxycodone and oxymorphone are metabolized in such different ways.
Source: Am J Manag Care. 2011;17:S276-S287
- It's not only Itraconazole
There are a large number of drugs that act as CYP inhibitors; all have the potential to artificially elevate the levels of any opioids that are metabolized by the CYPs. Here's a short list of some common drugs that inhibit CYP3A4 (including common beverages), and their relative strength of inhibition:
- Itraconazole and ketoconazole (strong)
- Ritonavir (one of the two drugs in Paxlovid) (strong)
- Erythromycin (moderate)
- Cimetidine (Tagamet) (weak)
- Certain herbal remedies and Green tea (strong)
- Grapefruit juice (moderate - strong depending on the brand)
A complete list can be found in many places, including here.
Bottom line
While there is nothing simple about drug-drug interactions with a little understanding it should become clear that these are not random events. Rather, they are logical and predictable. With our version of the "opioid wars" in full swing, knowledge is power. It is important for people taking these drugs to be aware that there are quite a few other drugs that will alter the normal processing of the opioid.
Addendum:
This same effect is found with other drugs, not just opioids. Here is a reference to a paper that discusses the impact of grapefruit juice on statin levels. Statin levels are significantly increased by one glass of grapefruit juice per day.
NOTES:
(1) Some drugs can induce rather than inhibit certain enzymes. This is less common and I have chosen not to cover it.
(2) There are more than 50 known CYPs, but only a handful of them are responsible for most drug metabolism. I doubt any of you are especially eager to hear about the other 45.

Josh Bloom
Director of Chemical and Pharmaceutical Science
Dr. Josh Bloom, the Director of Chemical and Pharmaceutical Science, comes from the world of drug discovery, where he did research for more than 20 years. He holds a Ph.D. in chemistry.


